

- Chromosomal microarray analysis (CMA), also known as array CGH is a diagnostic test that can detect clinically significant large (whole chromosome) and sub microscopic (microdeletion / microduplication) copy number changes throughout the genome.
- Chromosomal microarray analysis is the gold standard for the detection of large deletions and duplications along the whole genome. The higher the resolution and the K value of the test, the higher the sensitivity.
- This test allows the detection of microdeletions and microduplications of chromosome segments, which are too small to see under a microscope but may contain multiple genes.
CMA – Chromosomal Microarray

The Chromosomal MicroArray analysis test can be indicated across all life stages:
-
Preconception
CMA can be offered in the pre-conception phase if one or more of the below indications are present:
- If an affected child presents with a normal karyotype, but a genetic condition is suspected.
- If a child presents with an undiagnosed condition and the Whole Exome sequencing is negative.
- If an individual presents with undiagnosed intellectual or developmental delay that does not fit a specific syndrome (including Fragile X).
- To check whether a copy number variation that was detected in the affected child is de novo or inherited from their parents.
-
Prenatal
CMA is offered prenatally if one or more of the below indications are present:
- Abnormal fetal ultrasound
- Abnormal NIPT results indicated an increased risk of a chromosomally abnormal fetus
- Abnormal high-risk maternal serum screen
- The parents have a known chromosomal rearrangement, mosaicism or previous aneuploidies
- The parents have had previous livebirths or stillbirths with chromosomal abnormalities
- Fetal congenital abnormalities detected with ultrasound or MRI that indicate a significant risk of an unbalanced chromosomal abnormality
- Apparently balanced inherited rearrangements in a fetus with congenital abnormalities.
- Apparently balanced de novo rearrangements identified by G-band analysis (karyotype)
- High risk pregnancies
-
Post-natal
CMA is offered in childhood or adulthood if one or more of the below indications are present – the chromosomal microarray can be considered as a first line test in the following
- If an individual or fetus presents with multiple congenital anomalies that are not specific to a well identified genetic syndrome
- If a karyotype is negative and the individual’s phenotype is indicative of chromosomal aneuploidy.
- If an individual or fetus present with apparently nonsyndromic developmental delay or intellectual disabilities
- If an individual presents with autism spectrum disorders
- If a fetus is malformed or a still birth of unknown etiology occurs
The CMA can be used in cases where other tests have failed to yield a diagnosis specifically if one or more of the below symptoms or conditions are present
- Unexplained seizure disorder
- Growth delay
- Psychiatric illness
- Neuromuscular conditions
- Neuromuscular conditions
- Skeletal dysplasia
- Short stature
- Excessive growth
- Microcephaly
- Macrocephaly
-
Product of Conception
- CMA can be offered on a product of conception if there was a case of spontaneous abortions.
CMA can detect:
- Small chromosomal microdeletions and duplications
- Copy number variations
- Numerical chromosomal aneuploidy
- Unbalanced rearrangements
- Excessive homozygosity – platform dependent
- Suggestive risk of inheriting recessive disease or imprinting disorders – platform dependent
- Triploidy and tetraploidy – platform dependent
- Mosaicism greater than 20-30%
Two different types of CMA are available in Igenomix – CystoScan HD and CytoScan 750K. These two tests are similar but have different indications with CMA HD being the more sensitive test and CMA750K being the most cost effective.
CytoScan HD array
- Copy number probes (1.9 million) + SNP (750 K)
- Whole genome coverage
- Can detect regions of low heterozygosity, uniparental disomy (UPD), low level mosciasim and sample heterogeneity
- Highest probe density
CytoScan 750K
- Copy number probes only (550K) + SNP (200 K)
- Emphasis on clinically relevant regions
- Identification of regions of excessive homozygosity indicating UPD, may suggest consanguinity and determine candidate genes for further testing
Deletions smaller than 50 kb and duplications smaller than 400 kb may not be reviewed. Detected copy number variations (CNVs) are reported when found to have clear or suspected clinical relevance; CNVs devoid of relevant gene content or reported as common findings in the general population may not be reported. Regions of homozygosity are reported when a single LCSH is greater than 8-15 Mb (dependent upon chromosomal location and likelihood of imprinting disorder), or when the total autosomal LCSH proportion is greater than 3% (only autosomal LCSH greater than 3 Mb are considered for this estimate). Genomic linear positions are given relative to NCBI build 37 (hg19).
Test results are interpreted based on the recommendations and guidelines of International Standard of Cytogenomics Arrays (ISCA) as described below:
-
Positive (Pathogenic and likely pathogenic)
A positive result indicates that a copy number variant has been identified in association with the disease phenotype under study. This scenario will allow to provide genetic counselling or personal guidance regarding possible medical treatments, disease progression, reproductive-/prevention-strategies and potential implications for other family members.
-
Negative:
A negative result indicates that no disease-causing copy number variation was identified in the test performed. This does not guarantee that the induvial will be healthy or free from other genetic disorders or medical conditions. Additionally, a negative result does not rule out a genetic cause of the disease nor does it eliminate the risk for future offspring. However, if a negative test result is obtained and the variant in question is known to be present in affected family members, this then rules out a diagnosis of that genetic disorder in the proband. A negative result may be explained by several causes, including limited genetic knowledge and limitations associated to the used methodology.
-
Inconclusive / Variant of Uncertain Significance (VUS):
A finding of a variant of uncertain significance indicates that a copy number variation was detected, but it is currently unknown whether that CNV is associated with a genetic disorder or disease. A variant of uncertain significance is not the same as a positive result and does not clarify whether the proband is at an increased risk to develop a genetic disorder or disease. The change could be a normal genetic variant, or it could be disease-causing. Further analysis may be recommended, including testing both parents as well as other affected and unaffected family members. Sometimes, performing ancillary tests is necessary to prove the phenotype that the proband presents with. Detailed medical records or information from other family members also may be needed to help clarify the result.
Result interpretation is based on currently available information in the medical literature, research, and scientific databases. Because the literature, medical and scientific knowledge are constantly changing, new information that becomes available in the future may replace or add to the information that Igenomix used to interpret the results. Re-analysis of the results in previously issued reports considering new evidence is not routinely performed but is available upon request.
Sample Requirements
The following sample types are accepted for Igenomix genetic tests. A thorough labelling of the tube with unique identifying information is suggested, incorrect labelling can lead to rejection of the sample. The minimum required information to identify and accept a sample is – Patient’s full name, Date of birth, Gender and Medical Record Number.
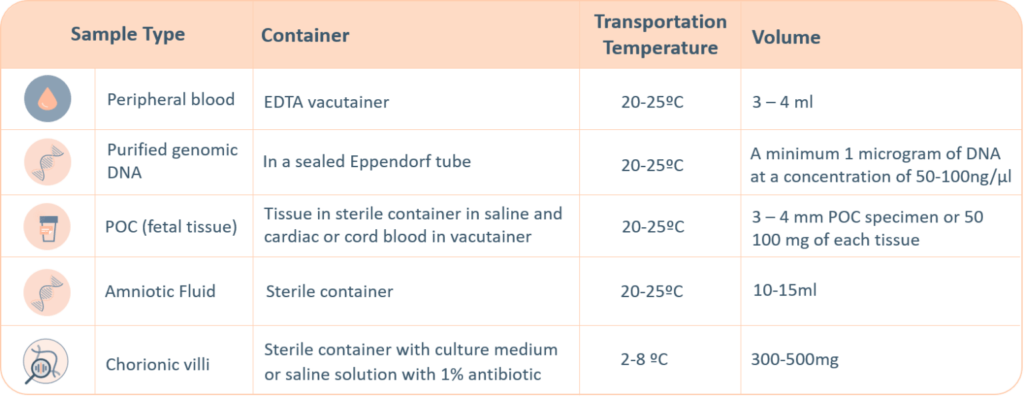
- Maternal blood sample must be sent with all products of conception, CVS and Amnio samples.
- Precedence will be given to all prenatal samples.
The ‘informed consent’ form and the ‘test requisition from’ (included within the provided kit) must be properly filled-in and signed by the patient and sent with the samples inside the shipping box or by e-mail to the laboratory. Igenomix will send you all the documents needed for the pick-up and transportation of the appropriate kit to our laboratory .
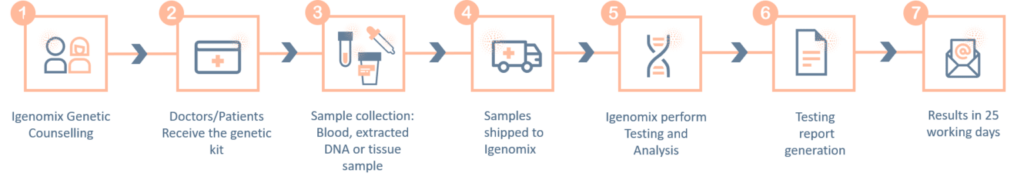
Methodology
Limitations
- CMA cannot identify:
- Balanced chromosomal rearrangements (balanced translocations, inversions)
- Small changes in the sequence of single genes (point mutations)
- Tiny duplications and deletions of DNA segments within a single gene (Fragile X syndrome, for example)
- Uniparental disomy (UPD)
- Methylation alterations
- Mosaicism less than 20%
- Complete ploidy

